A endocrinopediatra Mariana Zorron traz um conteúdo especial sobre um dos temas mais frequentes no consultório: a baixa estatura. Entenda a questão e atualize-se sobre diagnósticos e tratamentos.
Crescimento é um acontecimento exclusivo da criança e do adolescente, sendo um assunto muito particular ao pediatra. Embora ele seja dependente de fatores genéticos, fatores hormonais, psicológicos, sociais e nutricionais também atuam no crescimento. Fatores genéticos e hormonais possuem influência direta, enquanto que condições nutricionais fornecem os substratos plástico e energético, agindo juntamente com os fatores psicológicos na modulação dos fatores hormonais.
A baixa estatura é definida como estatura menor que “menos dois desvios-padrões” (-2DP), ou menor que percentil 3 para idade e sexo, ou ainda menor que o -2DP para o alvo parental.
Para uma adequada avaliação do crescimento, é necessária uma boa anamnese com antecedentes obstétricos, pessoais (trauma, infecções, irradiação), familiares (consanguinidade), além de um exame físico completo, com cálculo de velocidade de crescimento, medidas antropométricas e avaliação de estadiamento puberal.
O cálculo do alvo parental é realizado conforme a fórmula abaixo
(obs.: sempre medir a altura dos pais, pois as informações fornecidas podem estar equivocadas):
MENINAS = [ ALTURA MÃE + (ALTURA PAI − 13cm) ] ÷2 ± 8,5cm
MENINOS = [ ALTURA PAI + (ALTURA MÃE + 13cm) ] ÷2 ± 8,5cm
VELOCIDADE DE CRESCIMENTO DE CRIANÇAS E JOVENS
A velocidade de crescimento na infância varia conforme a faixa etária abaixo relacionada:
| VELOCIDADE DE CRESCIMENTO | |
| 1º ano | 25 cm/ano
•15 cm no 1º semestre •10 cm no 2º semestre |
| 2º ano | 10 cm/ano |
| 2 anos até o início da puberdade | 5–7 cm/ano |
| Puberdade | 9 cm/ano (meninas) |
| 10 cm/ano (meninos) | |
É sempre importante lembrar que, em meninas com baixa estatura, é essencial descartar a hipótese de Síndrome de Turner, mesmo que a criança não apresente sinais clínicos sugestivos.
O HORMÔNIO DO CRESCIMENTO (GH)
A secreção de GH ocorre de forma pulsátil, com picos de maior amplitude nas fases III e IV do sono. Geralmente ocorrem de 6 a 10 pulsos secretórios em 24 horas, principalmente no período noturno. A amplitude dos pulsos varia conforme a idade, aumentando na puberdade e decaindo na vida adulta.
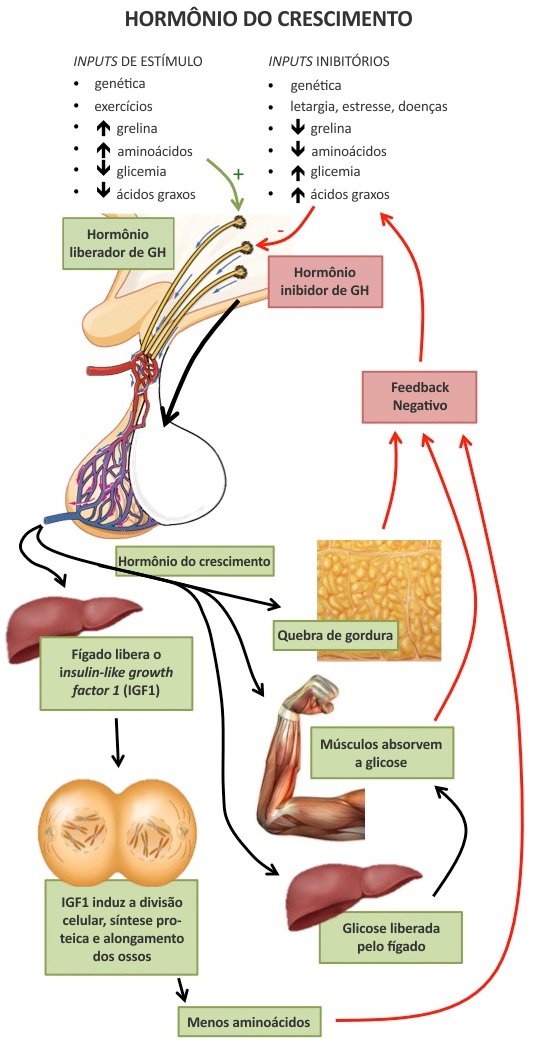
Os pulsos são controlados por um mecanismo complexo que envolve um controle hipotalâmico triplo (somatostatina, grelina e GHRH). A somatostatina determina o momento de ocorrência e amplitude dos pulsos, sem afetar sua biossíntese. O GHRH efetua a transcrição gênica e síntese do GH. A grelina estimula a secreção do GH. GHRH e somatostatina são influenciados por atividade física, nutrição, sono, estresse, esteroides sexuais e hormônios tireoidianos.
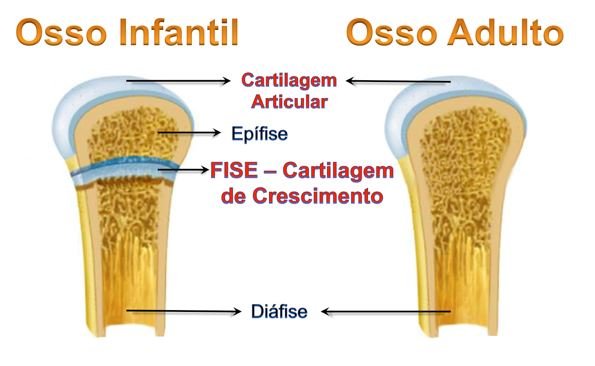
O crescimento linear se faz a partir da cartilagem de crescimento dos ossos longos, sendo esse crescimento mantido até o desaparecimento desta cartilagem.
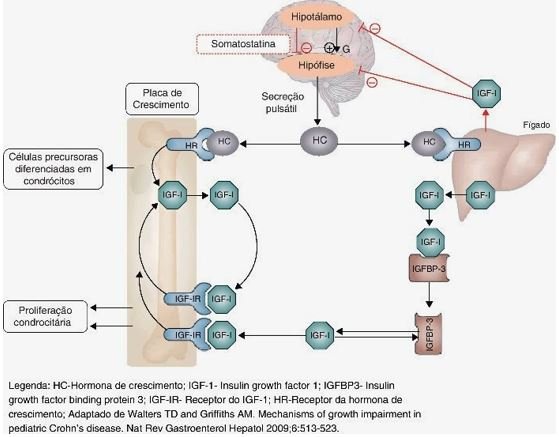
Os IGFs (insulin-like growth factors) são sintetizados principalmente pelo fígado, mas também por células ósseas e musculares, secundários ao estímulo de GH. Possuem ações insulina-símile, estimulando a síntese de DNA e proliferação celular.
Durante a fase intrauterina, os IGFs apresentam menor dependência em relação ao GH, que após o nascimento se torna o seu principal regulador. O IGF-1 encontra-se diminuído em restrições proteico-calóricas, sendo que, em casos de hiperalimentação, não ocorre sua elevação.
As IGFBPs (insulin-like growth factors binding proteins) são as proteínas ligadoras das IGFs, aumentando sua meia-vida e modulando suas ações em níveis autócrino, parácrino e endócrino.
CAUSAS DE DEFICIÊNCIA DE GH
- Processos inflamatórios/infecciosos (meningoencefalites, hipofisite autoimune);
- Processos infiltrativos (histiocitose, sarcoidose, hemossiderose);
- Alterações vasculares (aneurisma, infarto, anemia falciforme);
- Processos expansivos hipotalâmico-hipofisários (craniofaringioma, glioma, cisto, etc.);
- Sequela de tratamento de neoplasias (QTX/RTX até 10 anos após);
- Defeitos congênitos de linha média facial (displasia septo-óptica, fenda palatina, etc.);
- TCE (perinatais, acidentes, etc.);
- Genéticos (Sd. de Russel-Silver, Allagille, hipoplasia hipofisária, neuro-hipófise ectópica, etc.);
- Idiopática
DIAGNÓSTICO
- Exames laboratoriais e de imagem: RX de idade óssea, urina 1, gasometria, cálcio, fósforo, fosfatase alcalina, cariótipo (meninas), TSH, T4 livre, IGF-1, IGFBP-3, exames para descartar doenças crônicas conforme suspeita clínica (ex.: hemograma para anemia, provas de absorção intestinal para doenças disabsortivas, função renal para doenças renais, função hepática para doenças hepáticas etc.).
- GH basal não se presta como um exame de triagem pelo fato de ser um hormônio contrarregulador, com valores basais sem significado clínico. Para sua avaliação, são necessários testes dinâmicos.
A regulação do crescimento é complexa e multifatorial, sendo os fatores genéticos fundamentais para determinação do potencial do crescimento, porém com uma interface múltipla e articulada com os sistemas endócrino, imunológico e estado nutricional.
Dessa forma, o tratamento depende do diagnóstico etiológico. Por exemplo, se houver uma doença renal, hepática ou desnutrição, o tratamento deverá ser de acordo com a doença de base, reservando-se a reposição hormonal de GH aos casos abaixo descritos.

INDICAÇÕES DE REPOSIÇÃO DE GH PELA FDA (US Food and Drug Administration)
- Deficiência de GH isolada ou combinada
- Síndrome de Turner
- Insuficiência Renal Crônica
- Síndrome de Prader-Willi
- Criança pequena para a idade gestacional
- Baixa estatura idiopática
EFEITOS DO GH
- Hiperinsulinismo
- Aumento de lipólise (ativa a lipase lipoproteica)
- Estimula produção de IGF-1
- Induz a expansão clonal e a diferenciação de células precursoras da placa de crescimento
- Aumenta a reabsorção intestinal de fosfato e cálcio
- Anabolizante proteico
CONSENSO ATUAL
- Uso de doses terapêuticas não aumenta risco de neoplasias
- Aguardar no mínimo um ano após a cura de neoplasias para iniciar o uso do GH
- Evitar o uso em pacientes que apresentem doenças que por si só aumentem o risco de neoplasias (Sd. de Down, Sd. de Bloom, Fanconi etc.)
Compete ao pediatra geral verificar se a queixa de baixa estatura é real, através da medição de altura, que deverá ser plotada no gráfico de crescimento juntamente com o canal familiar, calcular a velocidade de crescimento, e realizar o diagnóstico diferencial de comorbidades crônicas que possam estar levando ao seu comprometimento antes de encaminhar ao especialista.
Lembrando que o crescimento é um processo dinâmico, portanto seu diagnóstico nunca é feito em uma única consulta.
REFERÊNCIAS
- Vieira, GK, Gonçalves, DF, Setian, N. In: Durval Damiani. (Org.). Endocrinologia na Prática Pediátrica. 2ed. Barueri: Manole, 2011, p. 31-50.
- Monte O, Longui CA, Calliari LE, Kochi C. Endocrinologia para o pediatra. 3. Ed. Rio de Janeiro: Atheneu, 2007.